OPLSAA Torsion Scan#
This example demonstrates loading a CHARMM molecule with the OPLSAA force field, performing a torsion scan around a bond, and computing energies and forces at each angle.
Imports#
[1]:
from collections import deque
from pathlib import Path
import jax.numpy as jnp
from jax import jit, grad
import matplotlib.pyplot as plt
import networkx as nx
import numpy as np
from jax_md.mm_forcefields import oplsaa
from jax_md.mm_forcefields.nonbonded.electrostatics import PMECoulomb
from jax_md.mm_forcefields.base import NonbondedOptions
DATA_DIR = Path(__file__).resolve().parent / 'data' / 'torsion-data' \
if '__file__' in dir() else Path('data/torsion-data')
Load CHARMM System#
[2]:
positions, topology, parameters = oplsaa.load_charmm_system(
str(DATA_DIR / 'scan_1.pdb'),
str(DATA_DIR / 'scan_1.prm'),
str(DATA_DIR / 'scan_1.rtf'),
)
for k, v in topology._asdict().items():
print(f"{k}: {f'shape={v.shape}' if hasattr(v, 'shape') else v}")
n_atoms: 22
bonds: shape=(23, 2)
angles: shape=(36, 3)
torsions: shape=(52, 4)
impropers: shape=(12, 4)
exclusion_mask: shape=(22, 22)
pair_14_mask: shape=(22, 22)
molecule_id: shape=(22,)
cmap_atoms: None
cmap_map_idx: None
exc_pairs: None
nbfix_atom_type: None
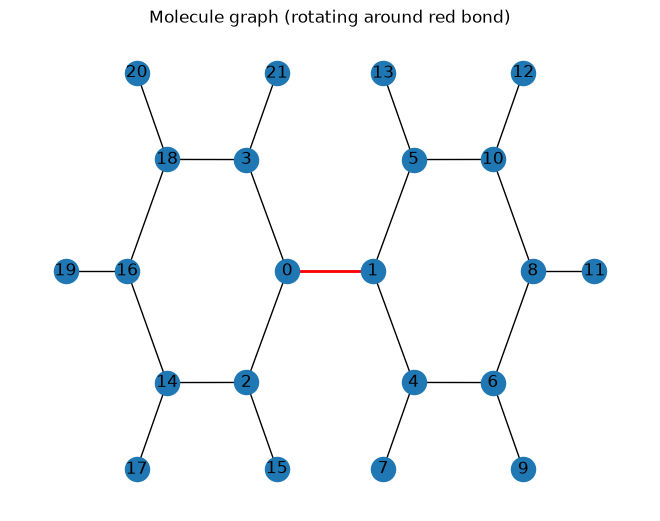
Visualize Molecule Graph#
[3]:
pos_2d = positions[:, :2]
bonds = topology.bonds
G = nx.Graph()
G.add_nodes_from(range(topology.n_atoms))
G.add_edges_from(bonds.tolist())
nx.draw(G, pos=pos_2d, with_labels=True)
nx.draw_networkx_edges(G, pos_2d, edgelist=[(0, 1)], edge_color='red', width=2)
plt.title('Molecule graph (rotating around red bond)')
plt.show()
Setup Energy Function#
Create the OPLSAA energy function with PME electrostatics.
[4]:
coords_range = jnp.max(positions, axis=0) - jnp.min(positions, axis=0)
box_size = coords_range + 20.0
box = jnp.array([box_size[0], box_size[1], box_size[2]])
nb_options = NonbondedOptions(
r_cut=12.0,
dr_threshold=0.5,
scale_14_lj=0.5,
scale_14_coul=0.5,
use_soft_lj=False,
use_shift_lj=False,
)
coulomb = PMECoulomb(grid_size=32, alpha=0.3, r_cut=12.0)
energy_fn, neighbor_fn, displacement_fn = oplsaa.energy(
topology, parameters, box, coulomb, nb_options
)
energy_fn_jit = jit(energy_fn)
[5]:
nlist = neighbor_fn.allocate(positions)
E_init = energy_fn_jit(positions, nlist)
for k, v in E_init.items():
print(f"{k}: {v}")
angle: 0.24455878138542175
bond: 1.4150346517562866
coulomb: -1.1449928283691406
improper: 0.0109158456325531
lj: 11.883728981018066
torsion: 0.0
total: 12.409245491027832
Torsion Scan Utilities#
[6]:
def find_bond_sides(bonds, bond_idx_to_break):
n_atoms = int(bonds.max()) + 1
bond_i, bond_j = bond_idx_to_break
adjacency = [set() for _ in range(n_atoms)]
for atom1, atom2 in bonds:
atom1, atom2 = atom1.item(), atom2.item()
if (atom1 == bond_i and atom2 == bond_j) or \
(atom1 == bond_j and atom2 == bond_i):
continue
adjacency[atom1].add(atom2)
adjacency[atom2].add(atom1)
def bfs(start):
side = set()
queue = deque([start])
visited = {start}
while queue:
atom = queue.popleft()
side.add(atom)
for neighbor in adjacency[atom]:
if neighbor not in visited:
visited.add(neighbor)
queue.append(neighbor)
return side
return bfs(bond_i), bfs(bond_j)
def set_dihedral_angle(pos, bonds, bond_atoms, target_angle_deg):
pos = jnp.array(pos)
i, j = bond_atoms
_, side2 = find_bond_sides(bonds, bond_atoms)
axis_vec = pos[j] - pos[i]
axis_vec = axis_vec / jnp.linalg.norm(axis_vec)
center = pos[i]
angle_rad = jnp.radians(target_angle_deg)
def rotate_point(p, axis, angle, center):
p_shifted = p - center
cos_a = jnp.cos(angle)
sin_a = jnp.sin(angle)
p_rot = (
p_shifted * cos_a
+ jnp.cross(axis, p_shifted) * sin_a
+ axis * jnp.dot(axis, p_shifted) * (1 - cos_a)
)
return p_rot + center
new_pos = pos.copy()
for atom_idx in side2:
if atom_idx != i:
new_pos = new_pos.at[atom_idx].set(
rotate_point(pos[atom_idx], axis_vec, angle_rad, center)
)
return new_pos
bonds_array = jnp.array(topology.bonds)
sidea, sideb = find_bond_sides(bonds_array, (0, 1))
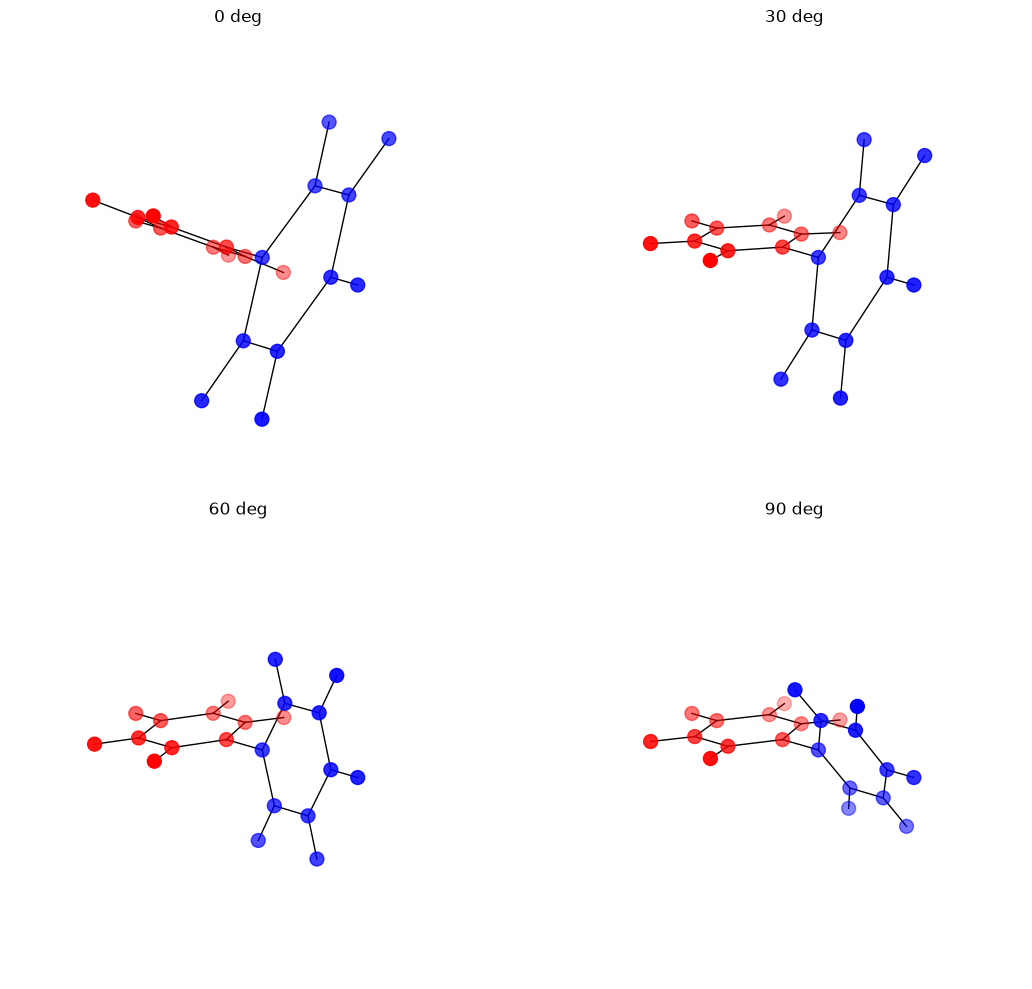
Visualize Rotated Conformations#
[7]:
fig = plt.figure(figsize=(12, 10))
angles_to_plot = jnp.linspace(0, 90, 4)
for idx, angle in enumerate(angles_to_plot, 1):
pos = set_dihedral_angle(positions, bonds_array, (0, 1), angle)
ax = fig.add_subplot(2, 2, idx, projection='3d')
colors = ['red' if i in sidea else 'blue' for i in range(len(pos))]
ax.scatter(*pos.T, c=colors, s=100)
for bond in bonds_array:
bi, bj = bond
ax.plot(
[pos[bi, 0], pos[bj, 0]],
[pos[bi, 1], pos[bj, 1]],
[pos[bi, 2], pos[bj, 2]], 'k-', linewidth=1,
)
ax.set_title(f'{angle:.0f} deg')
ax.set_axis_off()
plt.tight_layout()
plt.show()
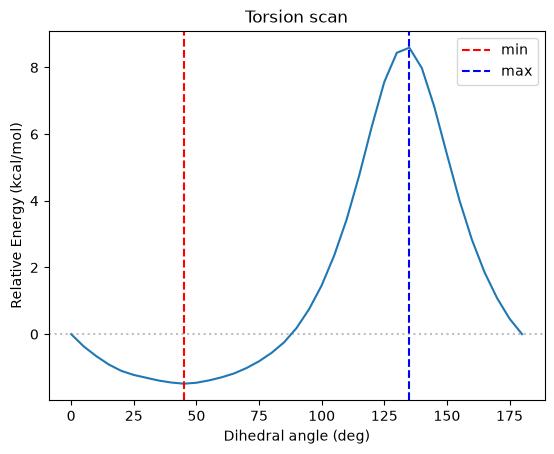
Torsion Scan#
[8]:
step = 5
angles_deg = jnp.arange(0, 181, step)
energies = []
nlist = neighbor_fn.allocate(positions)
print('Performing torsion scan...')
for angle_deg in angles_deg:
pos_rotated = set_dihedral_angle(positions, bonds_array, [0, 1], angle_deg)
pos_rotated_jax = jnp.array(pos_rotated)
nlist = neighbor_fn.update(pos_rotated_jax, nlist)
E = energy_fn_jit(pos_rotated_jax, nlist)
energies.append(float(E['total']))
if angle_deg % 30 == 0:
print(f" {angle_deg:3.0f} deg: E = {E['total']:>10.4f} kcal/mol")
energies = jnp.array(energies)
E_ref = energies[0]
rel_energies = energies - E_ref
e_argmin = rel_energies.argmin()
e_argmax = rel_energies.argmax()
print(f'\nScan complete!')
print(f'Min energy: {rel_energies[e_argmin]:.4f} kcal/mol at {angles_deg[e_argmin]:.0f} deg')
print(f'Max relative energy: {rel_energies[e_argmax]:.4f} kcal/mol at {angles_deg[e_argmax]:.0f} deg')
plt.figure()
plt.plot(angles_deg, rel_energies)
plt.xlabel('Dihedral angle (deg)')
plt.ylabel('Relative Energy (kcal/mol)')
plt.title('Torsion scan')
plt.axvline(angles_deg[e_argmin].item(), color='red', linestyle='--', label='min')
plt.axvline(angles_deg[e_argmax].item(), color='blue', linestyle='--', label='max')
plt.axhline(0, color='gray', linestyle=':', alpha=0.5)
plt.legend()
plt.show()
Performing torsion scan...
0 deg: E = 12.4092 kcal/mol
30 deg: E = 11.1009 kcal/mol
60 deg: E = 11.1132 kcal/mol
90 deg: E = 12.5957 kcal/mol
120 deg: E = 18.6353 kcal/mol
150 deg: E = 17.8069 kcal/mol
180 deg: E = 12.4092 kcal/mol
Scan complete!
Min energy: -1.4864 kcal/mol at 45 deg
Max relative energy: 8.5937 kcal/mol at 135 deg
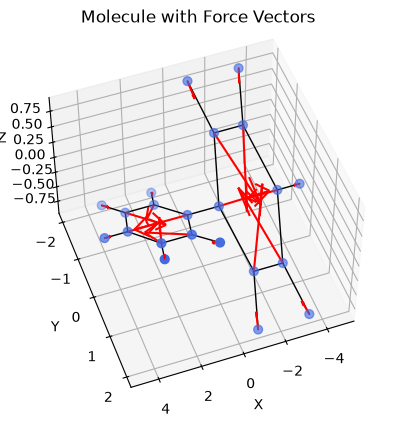
Forces#
[9]:
def e_total_func(pos, nlist):
return energy_fn_jit(pos, nlist)['total']
force_fn = jit(grad(e_total_func))
nlist = neighbor_fn.allocate(positions)
forces = force_fn(positions, nlist)
[10]:
fig = plt.figure()
ax = fig.add_subplot(111, projection='3d')
ax.scatter(positions[:, 0], positions[:, 1], positions[:, 2],
s=40, c='royalblue')
for bi, bj in bonds_array:
ax.plot(
[positions[bi, 0], positions[bj, 0]],
[positions[bi, 1], positions[bj, 1]],
[positions[bi, 2], positions[bj, 2]], 'k-', linewidth=1,
)
scale = 0.08
for p, f in zip(np.array(positions), np.array(forces)):
ax.quiver(p[0], p[1], p[2],
f[0] * scale, f[1] * scale, f[2] * scale,
color='red')
ax.set_xlabel('X')
ax.set_ylabel('Y')
ax.set_zlabel('Z')
ax.set_title('Molecule with Force Vectors')
ax.view_init(elev=50, azim=70)
plt.show()