Multi-Image Neighbor Lists for Small Periodic Boxes#
This tutorial demonstrates how to use multi-image neighbor lists in JAX-MD for systems where the cutoff radius is larger than half the box length (\(r_\text{cut} > L/2\)).
The Problem with Standard Neighbor Lists#
Standard neighbor lists in JAX-MD use the Minimum Image Convention (MIC), which assumes each atom interacts with at most one periodic image of every other atom. This works well when:
However, for small periodic boxes (common in ab initio MD or machine learning potentials with longer cutoffs), an atom may interact with multiple periodic images of the same neighbor. The multi-image neighbor list explicitly enumerates all images within the cutoff.
Imports & Setup#
[1]:
import os
IN_COLAB = 'COLAB_RELEASE_TAG' in os.environ
if IN_COLAB:
import subprocess
import sys
subprocess.run(
[
sys.executable,
'-m',
'pip',
'install',
'-q',
'git+https://github.com/jax-md/jax-md.git',
]
)
import numpy as onp
from jax import config
config.update('jax_enable_x64', True)
import jax.numpy as jnp
from jax import random, jit, lax
import time
import matplotlib.pyplot as plt
import seaborn as sns
from jax_md import space, energy, partition, quantity, simulate
from jax_md.custom_partition import neighbor_list_multi_image
from jax_md.custom_smap import pair_neighbor_list_multi_image
SMOKE_TEST = os.environ.get('READTHEDOCS', False)
sns.set_style(style='white')
def format_plot(x, y):
plt.xlabel(x, fontsize=20)
plt.ylabel(y, fontsize=20)
def finalize_plot(shape=(1, 1)):
plt.gcf().set_size_inches(
shape[0] * 1.5 * plt.gcf().get_size_inches()[1],
shape[1] * 1.5 * plt.gcf().get_size_inches()[1],
)
plt.tight_layout()
Matplotlib is building the font cache; this may take a moment.
Helper: Create Crystal Structures#
[2]:
def make_fcc(n_cells, a=1.0):
"""Create FCC crystal positions in fractional coordinates.
Args:
n_cells: Number of unit cells in each direction.
a: Lattice constant.
Returns:
R: Fractional positions of shape [N, 3] in [0, 1).
box: Box matrix of shape [3, 3] with columns as lattice vectors.
"""
# FCC basis: 4 atoms per unit cell
basis = onp.array(
[
[0.0, 0.0, 0.0],
[0.5, 0.5, 0.0],
[0.5, 0.0, 0.5],
[0.0, 0.5, 0.5],
]
)
positions = []
for i in range(n_cells):
for j in range(n_cells):
for k in range(n_cells):
for b in basis:
pos = (onp.array([i, j, k]) + b) / n_cells
positions.append(pos)
R = onp.array(positions)
L = n_cells * a
box = onp.eye(3) * L
return jnp.array(R), jnp.array(box)
def make_diamond_cubic(n_cells, a=5.43):
"""Create diamond cubic crystal using the 2-atom primitive cell.
Uses the FCC primitive cell with a 2-atom basis:
- Lattice vectors: a1=(0,1,1)a/2, a2=(1,0,1)a/2, a3=(1,1,0)a/2
- Basis: (0,0,0) and (1/4,1/4,1/4) in fractional coordinates
This is more efficient than the 8-atom conventional cell.
Used for silicon (a=5.43 Å) and germanium (a=5.66 Å).
Args:
n_cells: Number of primitive cells in each direction.
a: Conventional cubic lattice constant (default 5.43 Å for silicon).
Returns:
R: Fractional positions of shape [N, 3] in [0, 1).
box: Box matrix of shape [3, 3] with columns as FCC primitive vectors.
"""
# 2-atom basis in fractional coordinates of primitive cell
basis = onp.array(
[
[0.0, 0.0, 0.0],
[0.25, 0.25, 0.25],
]
)
positions = []
for i in range(n_cells):
for j in range(n_cells):
for k in range(n_cells):
for b in basis:
pos = (onp.array([i, j, k]) + b) / n_cells
positions.append(pos)
R = onp.array(positions)
# FCC primitive lattice vectors (columns of box matrix)
# a1 = (0, a/2, a/2), a2 = (a/2, 0, a/2), a3 = (a/2, a/2, 0)
box = (a / 2.0) * onp.array(
[
[0.0, 1.0, 1.0],
[1.0, 0.0, 1.0],
[1.0, 1.0, 0.0],
]
)
# Scale by n_cells
box = box * n_cells
return jnp.array(R), jnp.array(box)
Example 1: Lennard-Jones with All Three neighbor list formats#
We compute LJ energy for a small FCC argon crystal using all three neighbor list formats to verify they produce identical results:
Dense: Per-atom neighbor arrays
[N, max_neighbors]Sparse: Edge list
[2, capacity]with both directionsOrderedSparse: Edge list with one direction per pair (most efficient)
[3]:
# Argon LJ parameters (reduced units: sigma=1, epsilon=1)
sigma = 1.0 # Length unit
epsilon = 1.0 # Energy unit
r_cutoff = 2.5 * sigma
r_onset = 2.0 * sigma
# Create small FCC argon crystal where r_cut > L/2
# In reduced units, equilibrium nearest-neighbor distance ≈ 2^(1/6) * sigma ≈ 1.12
# FCC lattice constant a = sqrt(2) * nearest_neighbor ≈ 1.58 in reduced units
n_cells = 2
a_reduced = 1.55 # Small box to test multi-image (r_cut/L > 0.5)
R, box = make_fcc(n_cells, a=a_reduced)
N = len(R)
L = float(box[0, 0])
print(f'System: {N} Ar atoms in {n_cells}x{n_cells}x{n_cells} FCC')
print(f'Box size L = {L:.2f}sigma, r_cutoff = {r_cutoff:.2f}sigma')
print(f'r_cutoff / L = {r_cutoff / L:.2f} (> 0.5: multi-image needed)')
# Setup displacement function
displacement_fn, shift_fn = space.periodic_general(
box, fractional_coordinates=True
)
# Test all three formats
formats = [
('Dense', partition.Dense),
('Sparse', partition.Sparse),
('OrderedSparse', partition.OrderedSparse),
]
energies = {}
for name, fmt in formats:
neighbor_fn, energy_fn = energy.lennard_jones_neighbor_list(
displacement_fn,
box,
sigma=sigma,
epsilon=epsilon,
r_onset=r_onset / sigma,
r_cutoff=r_cutoff / sigma,
fractional_coordinates=True,
neighbor_list_fn=neighbor_list_multi_image,
pair_neighbor_list_fn=pair_neighbor_list_multi_image,
format=fmt,
)
nbrs = neighbor_fn.allocate(R)
E = float(energy_fn(R, nbrs))
energies[name] = E
# Get neighbor count
if partition.is_sparse(fmt):
n_edges = int(jnp.sum(nbrs.idx[0] < N))
else:
n_edges = int(jnp.sum(nbrs.idx < N))
print(f'{name:15s}: E = {E:12.6f}, edges = {n_edges}')
# Verify all formats give the same energy
E_ref = energies['Dense']
for name, E in energies.items():
assert abs(E - E_ref) < 1e-5, f'{name} energy mismatch: {E} vs {E_ref}'
System: 32 Ar atoms in 2x2x2 FCC
Box size L = 3.10sigma, r_cutoff = 2.50sigma
r_cutoff / L = 0.81 (> 0.5: multi-image needed)
Dense : E = -250.355417, edges = 4288
/home/docs/checkouts/readthedocs.org/user_builds/jax-md/envs/main/lib/python3.12/site-packages/jax/_src/ops/scatter.py:104: FutureWarning: scatter inputs have incompatible types: cannot safely cast value from dtype=int64 to dtype=int32 with jax_numpy_dtype_promotion=standard. In future JAX releases this will result in an error.
warnings.warn(
Sparse : E = -250.355417, edges = 4288
OrderedSparse : E = -250.355417, edges = 2144
Forces and Stress Computation#
We can compute forces using quantity.force and stress using quantity.stress. The multi-image neighbor list with graph_featurizer supports the perturbation kwarg required for stress calculation.
[4]:
# Use Sparse format for force/stress computation
neighbor_fn_lj, energy_fn_lj = energy.lennard_jones_neighbor_list(
displacement_fn,
box,
sigma=sigma,
epsilon=epsilon,
r_onset=r_onset / sigma,
r_cutoff=r_cutoff / sigma,
fractional_coordinates=True,
neighbor_list_fn=neighbor_list_multi_image,
pair_neighbor_list_fn=pair_neighbor_list_multi_image,
format=partition.Sparse,
)
# Perturb positions slightly from equilibrium to get non-zero forces
key = random.PRNGKey(42)
R_perturbed = R + random.normal(key, R.shape) * 0.01
nbrs_lj = neighbor_fn_lj.allocate(R_perturbed)
E_lj = float(energy_fn_lj(R_perturbed, nbrs_lj))
# Compute forces
force_fn = quantity.force(energy_fn_lj)
F = force_fn(R_perturbed, neighbor=nbrs_lj)
max_force = float(jnp.max(jnp.abs(F)))
print(f'Perturbed energy: {E_lj:.6f}')
print(f'Max force magnitude: {max_force:.6f}')
# Compute stress (3x3 tensor)
stress = quantity.stress(energy_fn_lj, R_perturbed, box, neighbor=nbrs_lj)
print(f'Stress tensor (diagonal): [{stress[0,0]:.4f}, {stress[1,1]:.4f}, {stress[2,2]:.4f}]')
print(f'Pressure: {-jnp.trace(stress) / 3:.6f}')
Perturbed energy: -232.244742
Max force magnitude: 51.575608
Stress tensor (diagonal): [2.8778, 3.3587, 3.1045]
Pressure: -3.113656
Example 2: Stillinger-Weber (Three-Body Potential)#
Stillinger-Weber is a three-body potential for silicon that requires Dense format for the angular terms. We use a 2x2x2 supercell of the 2-atom primitive cell.
Note: Stillinger-Weber internally uses space.map_neighbor for displacement computation, which applies the minimum image convention (MIC). For small boxes where r_cut > L/2, the multi-image neighbor list finds the correct neighbors, but the energy computation would still use MIC displacements. Therefore, we use a larger box where MIC is valid.
[5]:
# Stillinger-Weber parameters for silicon
sw_sigma = 2.0951 # Angstrom
sw_cutoff = 1.8 * sw_sigma # ~3.77 Angstrom
# Create 3x3x3 supercell so that MIC is valid
# For SW, the box must be large enough that r_cut < L/2
n_cells_sw = 3
a_sw = 5.43 # Si lattice constant
R_sw, box_sw = make_diamond_cubic(n_cells_sw, a=a_sw)
N_sw = len(R_sw)
# For non-cubic boxes, compute minimum perpendicular height
inv_box_T = jnp.linalg.inv(box_sw).T
heights_sw = 1.0 / jnp.linalg.norm(inv_box_T, axis=0)
L_min_sw = float(jnp.min(heights_sw))
print(f'System: {N_sw} Si atoms in {n_cells_sw}x{n_cells_sw}x{n_cells_sw} diamond cubic supercell')
print(f'Min box height = {L_min_sw:.2f} Angstrom, SW cutoff = {sw_cutoff:.2f} Angstrom')
print(f'cutoff / L_min = {sw_cutoff / L_min_sw:.2f} (< 0.5: MIC is valid)')
displacement_sw, shift_sw = space.periodic_general(
box_sw, fractional_coordinates=True
)
# Stillinger-Weber only supports Dense format (three-body terms)
# Note: SW uses MIC internally, so multi-image NL only helps with neighbor finding
neighbor_fn_sw, energy_fn_sw = energy.stillinger_weber_neighbor_list(
displacement_sw,
box_sw,
neighbor_list_fn=neighbor_list_multi_image,
format=partition.Dense,
fractional_coordinates=True,
)
nbrs_sw = neighbor_fn_sw.allocate(R_sw)
E_sw = float(energy_fn_sw(R_sw, nbrs_sw))
n_edges_sw = int(jnp.sum(nbrs_sw.idx < N_sw))
print(f'Stillinger-Weber energy: {E_sw:.6f} eV')
print(f'Number of edges: {n_edges_sw}')
print('Stillinger-Weber computes correctly (MIC valid for this box size).')
System: 54 Si atoms in 3x3x3 diamond cubic supercell
Min box height = 9.41 Angstrom, SW cutoff = 3.77 Angstrom
cutoff / L_min = 0.40 (< 0.5: MIC is valid)
Stillinger-Weber energy: -234.171986 eV
Number of edges: 864
Stillinger-Weber computes correctly (MIC valid for this box size).
Example 3: NVE Molecular Dynamics#
We run NVE (constant energy) molecular dynamics with the multi-image neighbor list. This demonstrates rebuild tracking and overflow handling following the pattern recommended in partition.neighbor_list documentation.
[6]:
# Simulation parameters
N_md = 500
dimension = 2
box_size = 40.0 if SMOKE_TEST else 60.0
# Create box matrix for 2D
box_md = jnp.eye(dimension) * box_size
# Random initial positions (fractional coordinates in [0, 1))
key = random.PRNGKey(0)
R_md = random.uniform(key, (N_md, dimension), minval=0.0, maxval=1.0)
# 50:50 mixture of two species
sigma_md = jnp.array([[1.0, 1.2], [1.2, 1.4]])
N_half = N_md // 2
species = jnp.where(jnp.arange(N_md) < N_half, 0, 1)
# Cutoff
r_cutoff_md = 2.5
print(f'System: {N_md} atoms in {dimension}D box of size {box_size}')
print(f'Cutoff: {r_cutoff_md}, cutoff/L = {r_cutoff_md / box_size:.3f}')
# Setup displacement function for fractional coordinates
displacement_md, shift_md = space.periodic_general(
box_md, fractional_coordinates=True
)
# For random positions, use generous capacity to avoid overflow
# Random positions can cluster, requiring more capacity than uniform estimates
# Use soft sphere potential with multi-image neighbor list
neighbor_fn_md, energy_fn_md = energy.soft_sphere_neighbor_list(
displacement_md,
box_md,
species=species,
sigma=sigma_md,
fractional_coordinates=True,
neighbor_list_fn=neighbor_list_multi_image,
pair_neighbor_list_fn=pair_neighbor_list_multi_image,
format=partition.Sparse,
)
# Initialize neighbor list
nbrs_md = neighbor_fn_md.allocate(R_md)
if nbrs_md.did_buffer_overflow:
raise RuntimeError('Neighbor list overflowed - increase max_neighbors')
# Setup NVE integrator
dt = 1e-2
init_fn, apply_fn = simulate.nve(energy_fn_md, shift_md, dt)
# Initialize state with zero temperature
state = init_fn(key, R_md, neighbor=nbrs_md, kT=0.0)
# JIT-compiled step function with neighbor list update
@jit
def step_fn(i, state_and_nbrs):
state, nbrs = state_and_nbrs
state = apply_fn(state, neighbor=nbrs)
nbrs = nbrs.update(state.position)
return state, nbrs
System: 500 atoms in 2D box of size 40.0
Cutoff: 2.5, cutoff/L = 0.062
[7]:
# Run simulation following JAX-MD's recommended pattern for overflow handling.
# See partition.neighbor_list docstring for the canonical example.
N_steps = 200 if SMOKE_TEST else 1000
print_every = 20
inner_steps = 10
PE = []
KE = []
rebuild_count = 0
realloc_count = 0
print(f'{"Step":>4} {"KE":>5} {"PE":>6} {"Total":>6} {"dt":>6} {"rebuild":>7} {"realloc":>7}')
old_time = time.time()
for i in range(N_steps):
# Track reference position before inner loop
old_ref_pos = nbrs_md.reference_position
# Run inner_steps using fori_loop for efficiency
new_state, new_nbrs = lax.fori_loop(0, inner_steps, step_fn, (state, nbrs_md))
# Check for buffer overflow after the loop
# If overflow: discard new state, reallocate with extra capacity
# If no overflow: accept new state
if new_nbrs.did_buffer_overflow:
# Reallocate with extra capacity (10 more neighbors per atom)
nbrs_md = neighbor_fn_md.allocate(state.position, extra_capacity=10)
realloc_count += 1
print(f' [Overflow at step {i * inner_steps}! Reallocating with extra capacity...]')
# Don't advance state - retry from last good state
else:
# Accept the new state
state = new_state
nbrs_md = new_nbrs
# Check if rebuild happened (reference position changed)
new_ref_pos = nbrs_md.reference_position
if not jnp.allclose(old_ref_pos, new_ref_pos):
rebuild_count += 1
pe = float(energy_fn_md(state.position, nbrs_md))
ke = float(quantity.kinetic_energy(momentum=state.momentum))
PE.append(pe)
KE.append(ke)
if i % print_every == 0 and i > 0:
new_time = time.time()
step_time = (new_time - old_time) / print_every / inner_steps
print(
f'{i * inner_steps:4d} {ke:5.1f} {pe:6.1f} {ke + pe:6.2f} '
f'{step_time:6.4f} {rebuild_count:7d} {realloc_count:7d}'
)
old_time = new_time
PE = jnp.array(PE)
KE = jnp.array(KE)
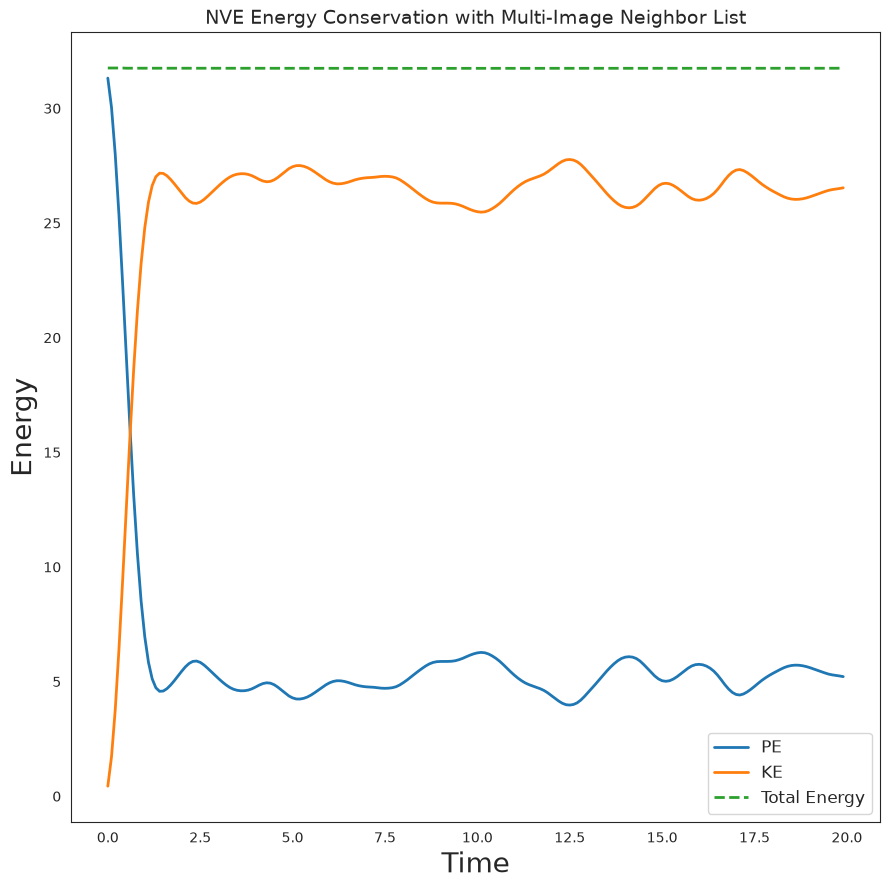
print(f'Total energy drift: {abs(float(PE[-1] + KE[-1] - PE[0] - KE[0])):.2e}')
print(f'Total rebuilds: {rebuild_count}, reallocs: {realloc_count}')
Step KE PE Total dt rebuild realloc
200 26.3 5.5 31.75 0.0223 16 0
400 27.0 4.8 31.75 0.0136 34 0
600 26.8 4.9 31.74 0.0141 51 0
800 26.8 5.0 31.74 0.0131 68 0
1000 25.5 6.3 31.74 0.0141 88 0
1200 27.4 4.4 31.74 0.0138 107 0
1400 25.7 6.1 31.75 0.0140 126 0
1600 26.0 5.8 31.75 0.0154 145 0
1800 26.4 5.4 31.75 0.0127 162 0
Total energy drift: 9.10e-03
Total rebuilds: 178, reallocs: 0
Plot Energy Evolution#
We verify energy conservation by plotting PE, KE, and total energy over time.
[8]:
t = onp.arange(N_steps) * dt * inner_steps
plt.figure(figsize=(10, 6))
plt.plot(t, PE, label='PE', linewidth=2)
plt.plot(t, KE, label='KE', linewidth=2)
plt.plot(t, PE + KE, label='Total Energy', linewidth=2, linestyle='--')
plt.legend(fontsize=12)
format_plot('Time', 'Energy')
plt.title('NVE Energy Conservation with Multi-Image Neighbor List', fontsize=14)
finalize_plot()
plt.savefig('nve_multi_image.png', dpi=150, bbox_inches='tight')
plt.show()
Visualize Final Configuration#
[9]:
ms = 40 if SMOKE_TEST else 15
R_final = onp.array(state.position)
# Convert from fractional to Cartesian for plotting
R_cart = R_final * box_size
plt.figure(figsize=(8, 8))
plt.plot(
R_cart[:N_half, 0], R_cart[:N_half, 1], 'o', markersize=ms * 0.5, alpha=0.7
)
plt.plot(
R_cart[N_half:, 0], R_cart[N_half:, 1], 'o', markersize=ms * 0.7, alpha=0.7
)
plt.xlim([0, box_size])
plt.ylim([0, box_size])
plt.axis('off')
plt.title('Final Configuration', fontsize=14)
finalize_plot((2, 2))
plt.savefig('nve_final_config.png', dpi=150, bbox_inches='tight')
plt.show()